Metagenomic data is a vast treasure trove of unknown proteins, potentially harboring novel CRISPR-Cas systems with diverse functions, waiting to be discovered.
However, traditional protein mining methods, such as BLAST or HMM, rely heavily on similarity comparisons with known sequences. This is like "looking for keys under a streetlight," making it difficult to discover truly novel protein families that differ significantly from known protein sequences.
On August 23, 2025, Nature Communications published the latest progress of a joint team led by Xiaokang Zhang, Jin Tang, Xingxu Huang, and Peixiang Ma, titled "Discovery of CRISPR-Cas12a clades using a large language model." This study leveraged an evolutionary-scale large-scale language model (ESM) to develop an AI-assisted scanning system called AIL-Scan. This system successfully transcends the constraints of sequence alignment and discerns key features within metagenomic data, discovering and identifying a series of previously unknown Cas12a isoforms and validating their unique potential in gene editing and molecular diagnostics.
Traditionally, protein function is determined by its three-dimensional structure, not just its amino acid sequence. Large-scale language models (LLMs), trained on massive amounts of protein sequences, can learn the structural and evolutionary information hidden within these sequences. Based on this concept, the research team constructed the AIL-Scan framework.
They first collected over 76,000 known Cas protein sequences from the NCBI database and fine-tuned ESM-2, a powerful protein language model, in a supervised manner. The trained model no longer relies on sequence alignment, but instead directly classifies Cas proteins by learning a deep "embedded" representation of the sequences.

Figure 1. Artificial Intelligence-assisted CRISPR-Cas Scan (AIL-Scan). (Feng, et al. 2025)
The model exhibited extremely high accuracy (98.22% accuracy for a 15 billion-parameter model) and strong generalization capabilities. In a new test set (TestSet2025) containing 3,601 Cas12 proteins, AIL-Scan successfully identified 3,182, while traditional HMM models only identified 1,240, demonstrating its powerful ability to discover new proteins.
Due to limited experimentally validated Cas protein data, directly predicting functions (such as trans-cleavage activity) using LLM is difficult. The team cleverly combined ESM embedding features with traditional machine learning models (such as LightGBM) to successfully construct a prediction model for Cas12a trans-cleavage activity on a small sample dataset, achieving an accuracy of 92.3%.
Based on the trained AIL-Scan, the research team scanned metagenomic databases, focusing on the widely used Cas12a family. Their findings overturned conventional understanding of the Cas12a CRISPR-Cas system. The classic Cas12a gene cluster typically consists of three integrases: Cas1, Cas2, and Cas4, responsible for integrating new spacer sequences. However, the research team discovered that many Cas12a gene clusters have different compositions. Based on the presence or absence of integrases, they classified the newly discovered Cas12a systems into eight new subtypes:
This classification is not only structural but also closely linked to function. For example, the absence of an integrase is significantly correlated with CRISPR gene cluster length and the number of spacers. Subtype VIII, lacking an integrase, has the shortest gene cluster and significantly fewer spacers.
So, what exactly are the differences between the newly discovered proteins? The research team selected 16 candidate Cas12a proteins from eight different subtypes and conducted comprehensive biochemical and cellular functional validation. Through high-throughput PAM screening, they discovered that these novel Cas12a proteins exhibit a wide range of PAM preferences. In addition to the classic T-rich PAMs (such as TTTV), they found that AmCas12a (from subtype II) prefers PAMs beginning with G, and RbrCas12a_1 (from subtype VII) recognizes the GTV PAM.
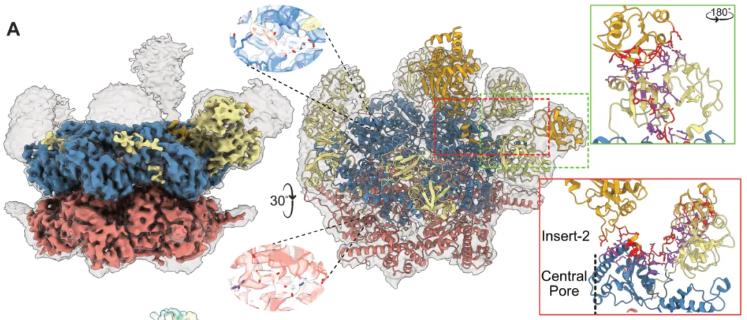
To explore the molecular basis for this diverse function, the researchers determined the cryo-electron microscopy structure of AmCas12a in complex with crRNA. The results revealed that the crRNA spacer region forms an unprecedented additional stem-loop structure, which may contribute to its unique function.
Furthermore, these novel Cas12a proteins exhibited significant differences in dsDNA cleavage activity, ssDNA trans-cleavage activity, and preference for different divalent metal ions.
In HEK293T cells, these novel Cas12a proteins also exhibited varying gene editing efficiencies, with AmCas12a, CAGCas12a, and the classic AsCas12a exhibiting higher editing activity.
The application of traditional Cas12a in molecular diagnostics is limited by its strict reliance on the TTTV PAM. Many important mutation sites, such as certain cancer SNPs, lack suitable PAMs near them, creating detection blind spots.
The research team successfully addressed this challenge by leveraging the newly discovered ability of AmCas12a to recognize a broad range of PAMs. Their study targeted KRAS G12C, a key cancer driver mutation whose surrounding sequences lack TTTV PAMs.
The results demonstrated that AmCas12a, guided by a specifically designed crRNA, efficiently recognizes and activates trans-cleavage activity, resulting in precise detection of the KRAS G12C mutation and lacks reactivity to wild-type sequences.
The detection system also exhibits exceptional sensitivity, capable of detecting as few as 10 copies of mutant DNA and accurately identifying a mutation abundance of 0.1% against a 1000-fold increase in wild-type background, surpassing the sensitivity of traditional Sanger sequencing.
In summary, this study successfully achieved "de-similarity" mining of novel Cas proteins by developing and applying the AIL-Scan strategy based on a large-scale language model, revealing unprecedented diversity within the Cas12a family. These newly discovered Cas12a proteins, with their unique biochemical properties (especially their diverse PAM recognition capabilities), significantly enrich the CRISPR toolbox, providing novel editing and diagnostic solutions for genomic sites inaccessible to traditional tools.
This work fully demonstrates the enormous potential of artificial intelligence in exploring uncharted territory in the life sciences and heralds a new era of AI-driven, function-driven protein discovery.
Get Latest Lifeasible News and Updates Directly to Your Inbox