On July 1, 2025, a team led by Longqing Sun from the Hubei Academy of Agricultural Sciences published a paper titled "Unraveling the regulatory network of barley grain metabolism through the integrative analysis of multiomics and mQTLs" in Nature Communications. This study addresses the still-unclear mechanisms governing metabolite abundance changes and gene expression during barley grain development by integrating multiomics and mQTL analysis.
The research team analyzed metabolomic and transcriptomic data from two barley varieties at six developmental stages, detecting numerous metabolites and co-expressed genes. They also identified metabolite-related mQTLs in a doubled haploid population and constructed a global co-expression regulatory network for barley grain metabolism. Furthermore, they identified key genes regulating the flavonoid metabolic pathway, revealing the regulatory mechanisms of grain color differentiation and providing important resources for improving barley nutritional quality.
The research team collected grain samples from Huadamai 6 (HB6) and Huaai 11 (HA11) at six key developmental stages (7, 14, 21, 28, 35, and 42 days after flowering) for metabolome analysis. High-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) detected 986 metabolites, 419 of which were annotated, encompassing amino acids and their derivatives, flavonoids, lipids, and other classes. Cluster analysis and principal component analysis (PCA) revealed that samples were clearly grouped by developmental stage, indicating stage-specific metabolite accumulation. Flavonoids were the most abundant of the annotated metabolites, accounting for 21.48%, while plant hormone derivatives and vitamins accounted for a lower proportion.
RNA-seq was performed on kernel samples from different developmental stages, yielding an average of 9.90 Gb of clean reads. Researchers identified 22,925 genes expressed in at least one developmental stage. PCA and cluster analysis of the transcriptome data also divided the samples into two groups based on developmental stage. Gene transcript levels within samples from the same developmental stage showed high correlation, similar to the results from the metabolome data. This suggests that both gene expression and metabolite accumulation patterns exhibit stage-specific characteristics during barley kernel development.
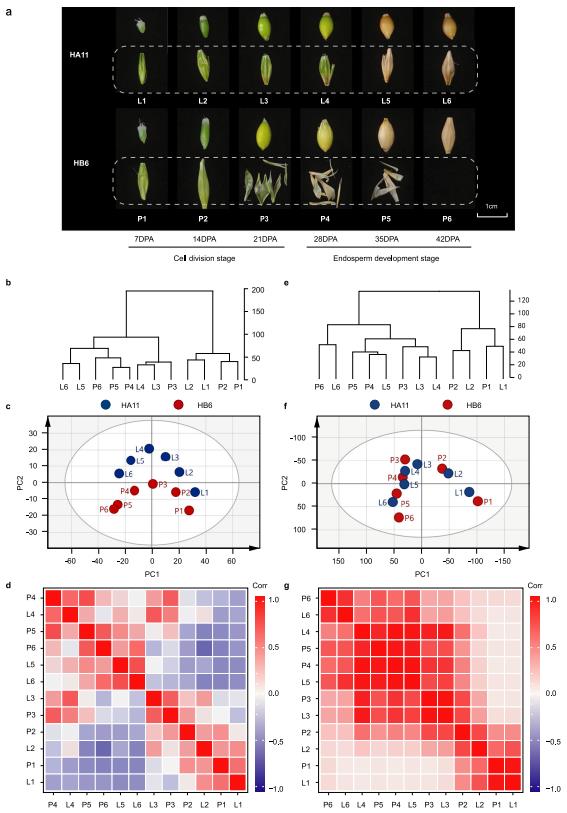
Figure 1. Summary of metabolome and transcriptome data sets. (Song, et al. 2025)
The k-means clustering algorithm was used to group 986 metabolites into nine clusters (MCs). Clusters 7 and 8 contained the most metabolites, with 261 and 147 metabolites, respectively. These metabolites primarily accumulate during early barley kernel development (7 DPA) and are gradually depleted after endosperm development (28 DPA). Metabolites in clusters 2 and 6 begin to accumulate after the onset of endosperm development, but peak at different times: cluster 2 reaches its peak at grain maturity (42 DPA), while cluster 6 reaches its peak at 35 DPA. Metabolites in cluster 4 are primarily produced at 28 and 35 DPA. These metabolites, which accumulate highly during endosperm development, may be associated with the nutritional quality of barley grains. Furthermore, the accumulation patterns and distribution of important active ingredients, such as flavonoids, amino acids, and their derivatives, are more diverse across clusters.
By calculating the Pearson correlation coefficient, 18,868 genes whose transcript levels were highly correlated with at least one metabolite accumulation pattern were identified and grouped into nine clusters (GCs). Results revealed a high degree of consistency between MC and GC. For example, Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis revealed that genes involved in amino acid metabolism pathways were enriched in GC2, while MC2 also contained a large number of amino acids, their derivatives, and lipid metabolites. Metabolic pathways such as flavonoid biosynthesis were enriched in GC5, while phenylpropanoid metabolites such as flavonoids were distributed in MC5. This suggests that the expression patterns of barley genes and the dynamic changes in metabolite accumulation are largely consistent.
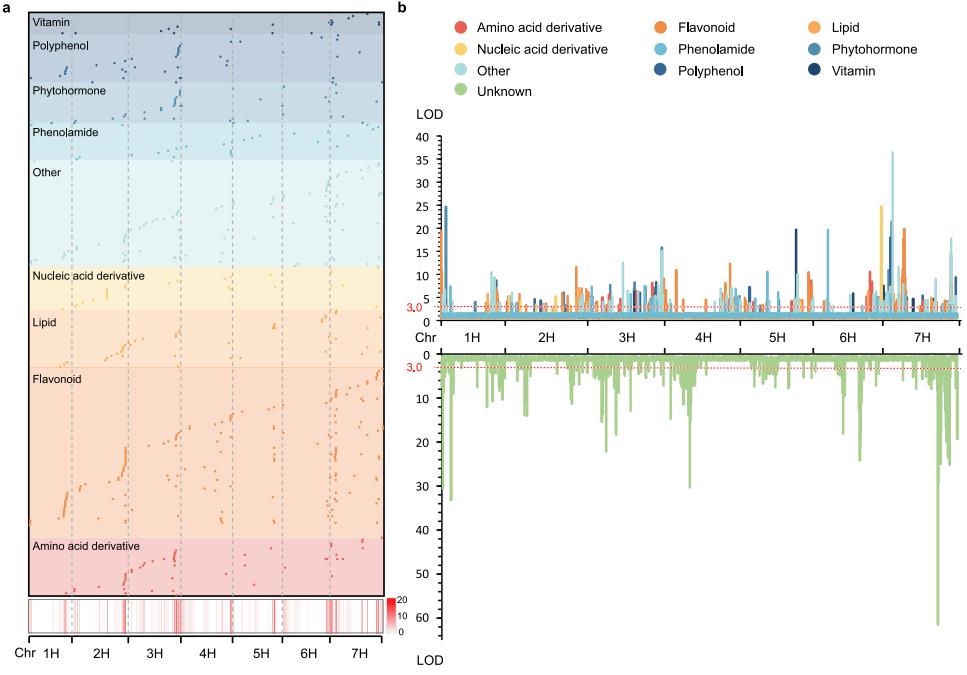
Figure 2. Chromosomal distribution of mQTL identified. (Song, et al. 2025)
Metabolite accumulation patterns differed between HB6 (hulled barley) and HA11 (naked barley) during grain development. Some clustered metabolites showed similar accumulation trends in both varieties, but at different levels. These differences may be related to differences in the transcriptional levels and gene functions of regulatory genes. For example, HA11 accumulates a large amount of glycosylated flavonoids, while the reported barley flavonoid glycosyltransferase gene in HB6 contains a base mutation, potentially affecting the structure and function of the encoded protein.
mQTL analysis of 1,150 metabolites detected in mature grains of a doubled haploid (DH) population identified 1,057 metabolite-related mQTL loci, 510 of which were for known metabolites, with flavonoid metabolites having the highest number (164). These mQTLs are primarily distributed on chromosomes 2H, 3H, and 7H, forming 43 mQTL hotspots. By identifying candidate genes within mQTLs, 265 candidate genes regulating metabolites were identified. By integrating mQTL information, candidate genes, and co-expression clustering, a global metabolic regulatory network (BMRN) for barley grain development was constructed.
In the BMRN, the dynamic accumulation patterns of 90 flavonoids during barley grain development and 164 genetic loci on barley chromosomes for 79 flavonoids were identified. Over 90% of flavonoids are glycosylated and primarily distributed in specific clusters. While some accumulate early in development, most accumulate during endosperm maturation. Flavonoid genetic loci are distributed across chromosomes, with high concentrations in hotspots.
Four genes regulating flavonoid glycosylation were identified (HvUGT-1, HvUGT-2, HvUGT-3, and HvUGT-4). In vitro enzyme activity analysis demonstrated their ability to glycosylate flavonoids. A series of structural genes involved in the flavonoid biosynthesis pathway, such as HvPAL-1 and Hv4CL-1, were also discovered. Transcription levels of these genes are high during endosperm maturation, and their homologous genes have been shown to be involved in the catalytic process of the flavonoid pathway in other crops. Furthermore, genes involved in the synthesis of precursors of the flavonoid metabolic pathway were discovered, and their expression is high during early grain development, indicating that flavonoid biosynthesis in barley grains is temporally regulated.
Two genes encoding R2R3-MYB transcription factors (HvMYB-1 and HvMYB-2) were identified in GC6. They are co-expressed with structural genes involved in flavonoid biosynthesis and flavonoid metabolites, and are expressed during endosperm development. Transient overexpression experiments showed that they upregulate the expression of structural genes involved in flavonoid biosynthesis in tobacco to varying degrees. Luciferase assays revealed that they regulate structural genes involved in the barley flavonoid pathway, indicating that these two transcription factors regulate flavonoid metabolism during barley grain development.
A gene encoding an NAC transcription factor (HvNAC-1) was found to be strongly co-expressed with the flavonoid metabolism pathway and shares many co-expressed genes and metabolites with HvMYB-1 and HvMYB-2. Transiently overexpressing HvNAC-1 in tobacco leaves significantly altered the accumulation levels of some flavonoid metabolites. Experiments confirmed that HvNAC-1 is localized to the nucleus and can induce the expression of structural genes involved in flavonoid biosynthesis, binding to CGTG binding elements in the promoters of these structural genes. Furthermore, the total flavonoid content and expression of key enzyme genes in leaves of HvNAC-1-overexpressing barley lines were significantly reduced. These results demonstrate for the first time that HvNAC-1 regulates the flavonoid metabolism pathway in barley.
Yeast two-hybrid, bimolecular fluorescence complementation, and co-immunoprecipitation experiments demonstrated a protein-protein interaction between HvMYB-1 and HvNAC-1. This discovery provides a potential transcriptional regulatory module for flavonoid biosynthesis beyond the traditional regulatory framework and provides a new understanding of the flavonoid biosynthesis pathway in barley grain metabolism.
Mature kernels of the barley DH population and its parents exhibit a certain degree of color differentiation. Data collected through sensory evaluation and color modeling, and PCA analysis demonstrated that these data effectively distinguish barley kernels of different color grades. Analysis revealed 17 metabolites significantly enriched in dark kernels, 14 of which were flavonoids, and 15 differentially expressed metabolites correlated with kernel color grade. Some flavonoids began to accumulate significantly during the middle and late stages of kernel development, consistent with kernel color formation, suggesting that flavonoid metabolites play an important role in kernel color formation.
QTL analysis for color-related parameters identified 35 QTLs on chromosomes 2, 3, 4, and 7. These QTLs co-localized with numerous flavonoid-related mQTLs, forming three QTL hotspot regions. The hotspot region contains a gene cluster (MbHF35), whose haplotypes are consistent with those reported to determine barley aleurone color. Two transcription factors (HvMYC-1 and HvC1-1) were also identified, with expression patterns and regulatory networks similar to those of known genes. Sequence analysis revealed differences between the two parents. Barley embryonic callus transformation experiments demonstrated that these factors are involved in grain color formation in the DH population, with their expression levels increasing as grain color deepens.
By integrating color-related QTLs into the global metabolic regulatory network, structural genes potentially involved in the anthocyanin pathway were identified. Experiments demonstrated that HvC1-1 and HvMYC-1 bind to the promoters of these structural genes and promote their expression. HvC1-1 may influence grain color formation by regulating the expression of HvMYC-1. Haplotype distribution analysis showed that kernel color is closely associated with the genotype composition of three loci (HvMYC-1, HvC1-1, and HvF35H). HvMYC-1 and HvC1-1 can, to a certain extent, compensate for the loss of related gene function, ensuring normal gene expression and resulting in darker kernel color.
This study comprehensively analyzed transcriptome and metabolome data from barley kernels at different developmental stages. The constructed dataset contains a large number of metabolites, expressed genes, and mQTL loci, providing rich information for in-depth analysis of the regulatory mechanisms of barley kernel development and improving nutritional quality.
Due to limitations in detection technology, some important metabolites were not detected, and some metabolites were not fully annotated. Future efforts, incorporating more detection technologies, updated databases, and new methods, will further refine the regulatory network and identify unknown metabolites. Furthermore, limited research exists on the regulation of barley flavonoid biosynthesis by miRNAs. Combining relevant databases with this study's database will hopefully establish a potential regulatory network.
This study constructed a global metabolic genetic regulatory network covering six distinct developmental stages of barley grains. This network clearly illustrates the dynamic accumulation patterns and transcriptional regulatory mechanisms of metabolites during barley grain development. Using this network, the study identified NAC transcription factors involved in flavonoid metabolism, further refining the flavonoid metabolic pathway during barley grain development and providing new insights into the genetic regulatory mechanisms of barley grain color formation. This study not only provides important insights into the regulatory mechanisms of other key metabolic pathways in barley grains but also provides a potential genetic resource for future nutritional quality improvement and breeding of barley.
Get Latest Lifeasible News and Updates Directly to Your Inbox