Understanding the genetic basis of important agronomic traits in crops is central to crop genetic improvement and precision breeding. Bulked Segregant Analysis (BSA) is a widely used gene mapping technique. It involves constructing two bulked pools of individuals exhibiting extreme phenotypes for a target trait (e.g., disease-resistant vs. disease-susceptible pools), and then genotyping these two pools to quickly identify genomic regions tightly linked to the trait, i.e., quantitative trait loci (QTLs). With the development of high-throughput sequencing (NGS) technology, the QTL-seq strategy, combining BSA with whole-genome sequencing, has greatly improved the efficiency and accuracy of mapping.
However, traditional BSA methods have a fundamental bottleneck: they are phenotype-driven. This means that researchers must first perform large-scale, precise phenotypic characterization of the entire segregating population, and then select individuals to construct bulked pools based on the phenotypic values of a specific trait. This process is not only labor-intensive, resource-intensive, and time-consuming, but the constructed DNA pools can usually only be used to analyze one trait. To study other traits, new bulked pools must be constructed, leading to inefficient utilization of data and resources. This inherent limitation severely restricts the application of the BSA method in synergistic improvement of multiple traits and the analysis of complex traits.
On January 12, 2026, a team led by Pasquale De Vita from the Council for Agricultural Research and Economics (CREA) published a paper in Plant Communications titled "Reverse BSA-QTLseq: a new genotype-driven bioinformatics approach for simultaneous trait mapping," proposing an innovative bioinformatics method that overturns the traditional BSA framework—Reverse BSA-QTLseq. This method shifts the core strategy from phenotype-driven to genotype-driven. By pre-sequencing individuals with the greatest genetic differences using high-throughput sequencing, and then performing virtual (in silico) pooled reconstruction and analysis of any number of traits on a computer, it achieves simultaneous and efficient QTL mapping for multiple traits in a single sequencing experiment.
The logic of the traditional BSA method is "phenotypic difference to genotypic difference," while Reverse BSA-QTLseq reverses this to "genotypic difference to phenotypic difference." The core idea is that the combination of individuals with the greatest genetic distance in a population naturally contains most of the variation information for the traits in that population. Therefore, by pre-selecting and sequencing these genetically most representative individuals, a reusable digital gene bank containing the genetic information of the entire population is established. Subsequently, researchers can perform phenotypic characterization of this core group for any number of traits, and based on the phenotypic values of different traits, flexibly and repeatedly reconstruct different extreme mixed pools on a computer for QTL analysis.
The specific workflow of this method is divided into two steps:
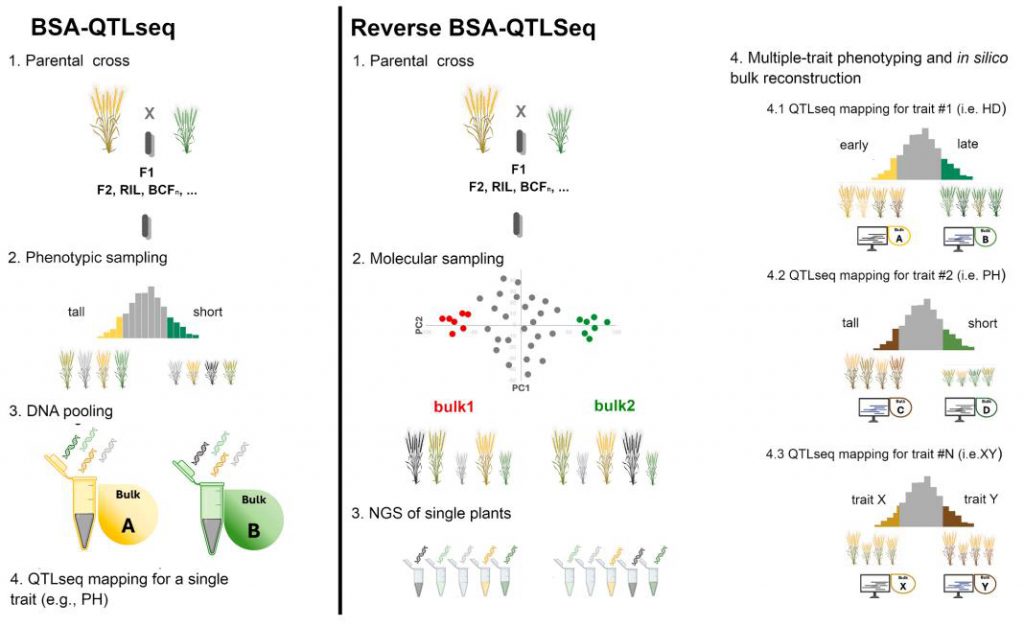
Figure 1. Working model. (Esposito, et al.2025)
To validate the effectiveness of the new method, the research team analyzed two key agronomic traits, plant height (PH) and heading date (HD), using a common wheat RIL population. They first used SNP chip data and PCA analysis to select 46 families with the greatest genetic distance from 176 RILs (plus two parents) as the core population. Subsequently, whole-exome sequencing was performed on these 48 individuals.
After obtaining the phenotypic data for plant height and heading date from these 48 individuals, the researchers constructed high-plant/short-plant mixed pools and early-flowering/late-flowering mixed pools in silico. Through QTL-seq analysis, they successfully identified 28 QTLs for plant height and 30 QTLs for heading date. These localization results were highly consistent with several known major genes. For example, the plant height QTLs were precisely located in the regions containing the famous dwarf genes Rht-B1 (on chromosome 4B) and Rht-5 (on chromosome 3B); the heading date QTLs accurately targeted the core gene Vrn-A1 (on chromosome 5A), which controls vernalization and flowering time. In addition, the study also discovered several new, previously unreported QTL loci, demonstrating the powerful QTL discovery capability of this method.
To further screen candidate genes from the QTL regions, the research team performed transcriptome sequencing on the two parental lines of the RIL population and analyzed differentially expressed genes (DEGs) in roots and leaves. By integrating QTL mapping results, variant site annotation information, and DEG data between parents, they successfully identified a set of high-confidence candidate genes for plant height and heading date. The functions of these genes are involved in multiple biological processes, including photoperiod regulation, hormone signaling, nutrient transport, and stress response.
To experimentally validate the function of the candidate genes, the researchers used a wheat TILLING mutant library to find a nonsense mutant of a candidate gene (an F-box protein gene) located in the plant height QTL region on chromosome 2B. Phenotypic analysis confirmed that the mutant plants showed a significant reduction in plant height of 14 cm, thus strongly validating the function of this gene in regulating wheat plant height and demonstrating the effectiveness of Reverse BSA-QTLseq combined with multi-omics analysis for gene discovery.
To test whether Reverse BSA-QTLseq is applicable to other species and different traits, the research team used a publicly available pea RIL population dataset to conduct an independent validation analysis of resistance to Striga (a parasitic weed). They compared the mapping performance of Reverse BSA-QTLseq with traditional BSA-QTLseq under different pool sizes (5, 10, and 25 individuals).
The results showed that Reverse BSA-QTLseq performed significantly better than the traditional method. With a pool size of 10 individuals, the new method not only successfully mapped all three known major resistance QTLs with higher resolution and signal-to-noise ratio, but also identified several minor QTLs that the traditional method failed to detect. In contrast, the traditional BSA method showed weaker signals, lower mapping accuracy, and was more susceptible to noise interference. This result fully demonstrates that the Reverse BSA-QTLseq method has good universality and that the genotype pre-selection strategy effectively improves the statistical power and accuracy of QTL mapping.
This study successfully developed and validated an innovative gene mapping strategy called Reverse BSA-QTLseq.
This method fundamentally addresses the core pain points of traditional methods—time-consuming, labor-intensive, poor flexibility, and inability to reuse data—by transforming the phenotype-driven paradigm of traditional BSA into a genotype-driven approach. Through two successful case studies in wheat and peas, the study systematically demonstrated the following outstanding advantages of the new method:
The development of the Reverse BSA-QTLseq method provides a powerful new tool for crop functional genomics research and molecular breeding practices. It not only accelerates the discovery of genes controlling complex agronomic traits but also provides a feasible technical path for multi-trait synergistic improvement in variable environments. In the future, combining this framework with more omics technologies (such as transcriptomics and epigenomics) is expected to reveal the complex regulatory network between genotype and phenotype more deeply, thus promoting crop breeding into a more precise and efficient era of intelligent breeding.
Get Latest Lifeasible News and Updates Directly to Your Inbox
Structural Factors Affecting CRISPR-Cas9 Trans-Cleavage Activity
February 9, 2026
The Dual Identity of Wheat Powdery Mildew Effector Proteins
February 3, 2026