On June 26, 2025, a team led by Richard A. Nichols from Queen Mary University of London published a research paper in Science titled "Rapid polygenic adaptation in a wild population of ash trees under a novel fungal epidemic." This study is the first to demonstrate in a natural population that European ash trees (Fraxinus excelsior), when faced with infection by the invasive pathogenic fungus Hymenoscyphus fraxineus, achieved heritable disease resistance evolution in just one generation through the coordinated changes in allele frequencies at thousands of small-effect gene loci. This finding provides crucial empirical evidence for the theory of polygenic adaptive evolution, revealing the rapid evolutionary mechanisms by which natural populations respond to emerging diseases, and has landmark significance for understanding biological adaptive evolution and the conservation of endangered species.

It remains an unsolved mystery in evolutionary biology whether complex traits adapt to new environments through large changes in allele frequencies at a few gene loci or through small changes at many loci.
Theory suggests that highly polygenic responses should be both rapid and effective, but in nature, population geneticists more readily find evidence of natural selection involving a few large-effect loci.
While quantitative genetics methods can demonstrate additive genetic variation in complex traits under selection through statistical comparisons of related individuals, these methods often struggle to prove that a selection response is actually occurring. This has led to a disconnect between population genetics and quantitative genetics.
The authors propose using genomic prediction methods, originally developed for agricultural breeding, to predict the genetic value of individuals for a quantitative trait based on whole-genome single nucleotide polymorphism (SNP) data, to bridge this gap.
If the authors can observe significant differences in whole-genome allele frequencies before and after the emergence of new selective pressures, or across different age groups at the same time point, and these changes affect genetic value, then a polygenic selection response can be demonstrated.
In the ash dieback epidemic that has swept across Europe over the past thirty years, the invasive pathogen Hymenoscyphus fraxineus, originating from East Asia, has subjected ash tree populations to strong selective pressure.
Several studies based on planting trials have shown that there is genetic variation in susceptibility to this fungus within European ash populations.
The selective pressure may be stronger on smaller, younger ash trees, as they are observed to die more readily from dieback infection than larger, older trees.
The rapid death of young trees may be due to the fungus being able to encircle the main stem more quickly, and because smaller trees are closer to the leaf litter layer where fungal fruiting bodies are produced. The replenishment of the next generation may also be affected by reduced reproductive capacity in adult trees that are damaged but not killed.
It is hypothesized that the death of susceptible young trees and reduced reproduction in susceptible adult trees will drive changes in allele frequencies, thereby increasing disease resistance in subsequent generations of ash trees.
In a previous study, the authors sequenced nearly 38,000 ash trees, approximately 7 years old, from the UK, Ireland, and Germany. They selected 623 trees with the best health status (highest rating) and 627 trees with severe damage to their woody tissue from ash dieback disease (lower rating). Using genome-wide association studies (GWAS), the authors ranked gene loci associated with these phenotypes by significance and constructed genomic prediction models using the top 100 to 50,000 loci, testing them on 148 trees.
The results showed that the model using approximately 10,000 loci had the highest correct classification rate for the test sample. The authors calculated that these genomic estimated breeding values (GEBVs) explained approximately 24% of the phenotypic variation in ash dieback disease in the test population.
If natural selection is indeed acting on natural ash tree populations under high disease pressure, the team of Richard J. A. Buggs/Richard A. Nichols at Queen Mary University of London expected to see an increase in GEBVs in young tree generations infected from seed germination, with changes in allele frequencies correlating with their effect size. This study tested this hypothesis in a woodland located within the geographical range of the aforementioned sample sites.
The authors' research site, Marden Park woodland, is an ancient semi-natural woodland primarily composed of European ash trees, where H. fraxineus has been present since 2012.
In 2019, the authors phenotypically assessed the trees in this area and collected samples for sequencing, finding that most trees showed disease symptoms, with no evidence of felling or removal of dead trees.
By 2021, disease damage, especially in young trees, had further intensified, consistent with widespread reports of ash dieback epidemics across Europe.
The authors performed short-read sequencing with approximately 11x genome coverage on 580 individuals (including 128 mature trees before the epidemic and 452 young trees grown after the epidemic) and 30 technical replicate samples, estimating allele frequencies at approximately 9 million SNP loci.
Of the 10,000 SNPs previously used for genomic prediction, 7985 loci showed polymorphism in the Marden Park population and met quality standards.
Compared to planting trials including seeds from multiple origins, the Marden Park population had lower genetic diversity, resulting in 2015 loci not showing polymorphism, some fixed to alleles associated with low disease incidence, and others fixed to alleles associated with high disease incidence.
The authors calculated the GEBVs of the Marden Park trees using these 7985 loci based on genomic prediction model parameters trained on planting trials.
The authors scored disease in young trees using a five-point scale and assessed mature trees by canopy cover percentage, finding no significant correlation between individual GEBVs and field-assessed disease phenotypes.
Given that field disease phenotypes are heavily influenced by local environment, age, and other microorganisms, and that different assessment methods were used for mature and young trees, this weak correlation was expected.
Mature tree canopy cover may not accurately reflect their genetic resistance to fungal damage during their juvenile stage.
Under conditions of significant environmental influence, the breeding value of mature trees is better estimated through their offspring phenotypes rather than their own phenotypes.
Using parentage inference based on 1000 SNPs, the authors found a significant correlation between parental GEBVs and the average health scores of their offspring in 36 mature trees and their 121 offspring, indicating that the authors' GEBVs effectively predicted genetic resistance at the juvenile stage; however, there was no significant correlation between parental visual health scores and their offspring health scores.
In summary, these results support the idea that polygenic inherited disease resistance is at play in natural ash tree populations.
Get Latest Lifeasible News and Updates Directly to Your Inbox
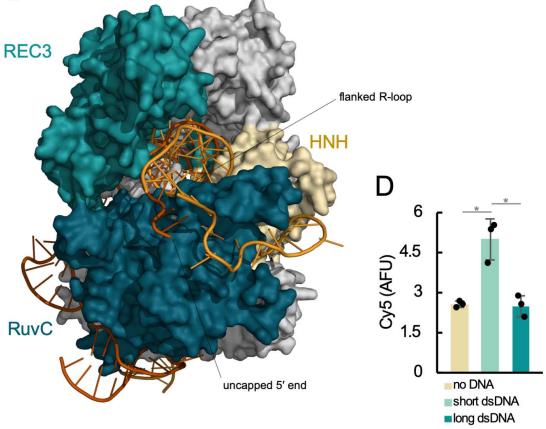
Structural Factors Affecting CRISPR-Cas9 Trans-Cleavage Activity
February 9, 2026
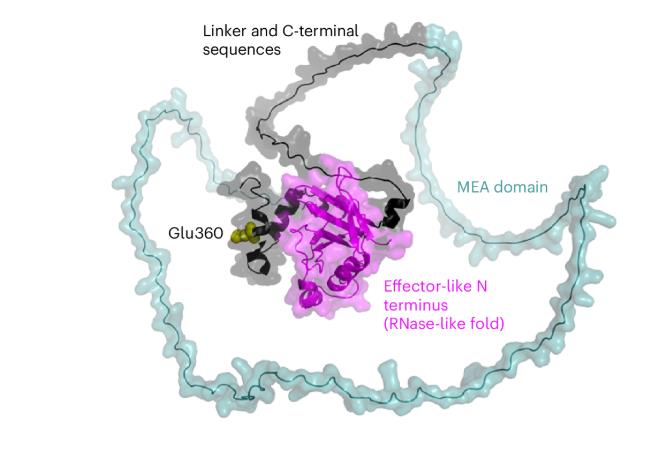
The Dual Identity of Wheat Powdery Mildew Effector Proteins
February 3, 2026