The CRISPR-Cas9 system has become a cornerstone of modern molecular biology, powering applications from gene knockout to base editing across diverse organisms. Yet the repertoire of Cas9 proteins available for practical use remains remarkably small. Most Cas9 variants in wide use today — SpCas9, SaCas9, Nme2Cas9, and a handful of others — were discovered through homology-based or similarity-driven mining strategies that rely on multiple rounds of BLAST searches and subjective parameter tuning.
This dependence on sequence alignment introduces several bottlenecks:
Here lies a critical insight: Cas9 proteins can differ dramatically in amino acid sequence while performing essentially the same DNA recognition and cleavage functions. This suggests that important determinants of Cas9 identity exist beyond the handful of conserved domains that homology searches rely on. Deep learning, with its capacity to learn latent features from sequence data without explicit annotation, offers a natural solution to this problem. While neural networks have already been applied to predict gRNA efficiency and off-target rates, their potential for de novo Cas9 protein mining had remained largely unexplored — until now.
The developers of CasMiner constructed their training datasets from UniRef90, extracting 1,946 protein sequences annotated as Cas9 nucleases (801–1,820 amino acids) as the positive set. For the negative set, they employed a shuffling strategy: each positive sequence was randomly permuted at varying thresholds (10% to 100%, in 10% increments), preserving amino acid composition while destroying the hidden sequence features that define Cas9 function. This produced 10 distinct negative datasets, each paired with the same positive set to train a separate model.
Each of the 10 models uses a CNN-LSTM architecture. The convolutional layers capture local sequence motifs, while the LSTM layers model longer-range dependencies. Among the 10 models, the one trained on Dataset80 — where negative sequences were 80% shuffled — delivered the best performance:
A classifier is only useful if it can distinguish Cas9 from proteins that share functional similarities. CasMiner was tested against several challenging negative sets:
These results confirm that CasMiner does not simply learn surface-level sequence statistics — it captures features specific to Cas9 that are not shared by other nuclease families.
Using Grad-CAM to extract features from the convolutional layers, the researchers identified a distinctive activation pattern in Cas9 sequences that they termed the Cas9 fingerprint. Non-Cas9 sequences showed no such pattern. Each fingerprint contains two prominent peaks corresponding to active-site residues — for example, D10 and H840 in SpCas9 — and most also exhibit peaks associated with DNA-binding residues such as H983 in SpCas9 and H701 in SaCas9. This internal representation demonstrates that CasMiner learns biologically meaningful features rather than statistical artifacts.
Armed with CasMiner, the team turned to the proGenomes non-redundant genome collection. They pre-filtered proteins to the 800–1,600 residue range, subdivided them into eight subsets by 100-amino-acid increments, and used Pfam domain annotation to prioritize the subset most enriched for Cas9-associated domains. The 1,301–1,400 AA subset contained the most RuvC_III, Cas9_BH, Cas9_REC, HNH_4, and Cas9_PI domain annotations.
CasMiner scanned this subset and identified 333 sequences with prediction scores above 50%, of which 262 scored at or above 90%. After downloading the corresponding 214 genomes and recovering 218 candidate sequences, the researchers used the CRISPR Recognition Tool to associate candidates with CRISPR repeat arrays. The top-scoring candidate lacked associated repeats, so the second-highest candidate — linked to 56 repeats in the genome of Vagococcus penaei CD276T — was selected and named VpCas9.
VpCas9 recognizes the PAM NGGV (N: A/T/C/G; V: G/T). The first three positions, NGG, match SpCas9, while the fourth position prefers G and tolerates T. Cleavage occurs 3–4 nucleotides upstream of the PAM, consistent with SpCas9.
In vitro cleavage assays confirmed that VpCas9 robustly cuts substrate DNA. Overall cleavage rates were 75.52% on target strands and 76.91% on non-target strands.
An E. coli fluorescence reporter system provided further evidence:
AlphaFold3 predicted structures revealed an RMSD of only 1.508 Å between VpCas9 and SpCas9. A notable structural difference is that Vp-sgRNA's Stem-loop 3 forms a taller, narrower structure. VpCas9 accommodates this through a groove formed by residues E726 and R729, while SpCas9 uses a flatter surface (D718 and H721) that can accept both sgRNA scaffolds.
The same deep-learning features that enable Cas9 identification can also guide protein engineering. The researchers retrieved 851 VpCas9 homologs from UniRef90, extracted their feature matrices, and computed a core-site matrix and a position-specific amino acid probability (PSAP) conservation matrix. By calculating the difference (Diff) between mutation and wild-type residues across both matrices, they ranked candidate mutations and selected 12 target sites with total ranking scores below 30.
A qPCR-based method was developed to quantify editing efficiency. Among the 12 single mutants, 9 outperformed wild-type VpCas9, with V623I showing the highest efficiency. Building on this result, the team generated three double mutants on the V623I background:
All three double mutants achieved significantly higher editing efficiency than wild-type VpCas9.
Molecular dynamics simulations over 500 ns revealed that all three mutants exhibit reduced fluctuations in the HNH domain, indicating increased structural rigidity. VPM2-1, VPM2-2, and VPM2-3 also showed dampened fluctuations in the PI and REC2 domains. Notably, all mutation sites reside in the REC1_2 domain — distant from the HNH and PI domains — suggesting that these substitutions modulate protein dynamics through long-range allosteric effects.
Surface electrostatic potential analysis showed that all mutants carry more positive charge than wild-type VpCas9:
The added positive charge concentrates at the entrance of the HNH-REC1 channel or along the inner wall of the PI-REC1 cavity, enhancing electrostatic attraction to the negatively charged DNA target strand.
Principal component analysis and free energy landscape calculations further demonstrated that the mutants explore a narrower conformational space with broader minimum free energy basins — VpCas9: 315.37 Å2; VPM2-2: 1,050.41 Å2; VPM2-3: 987.88 Å2 — implying that the mutants adopt more stable conformations favorable for DNA binding and cleavage.
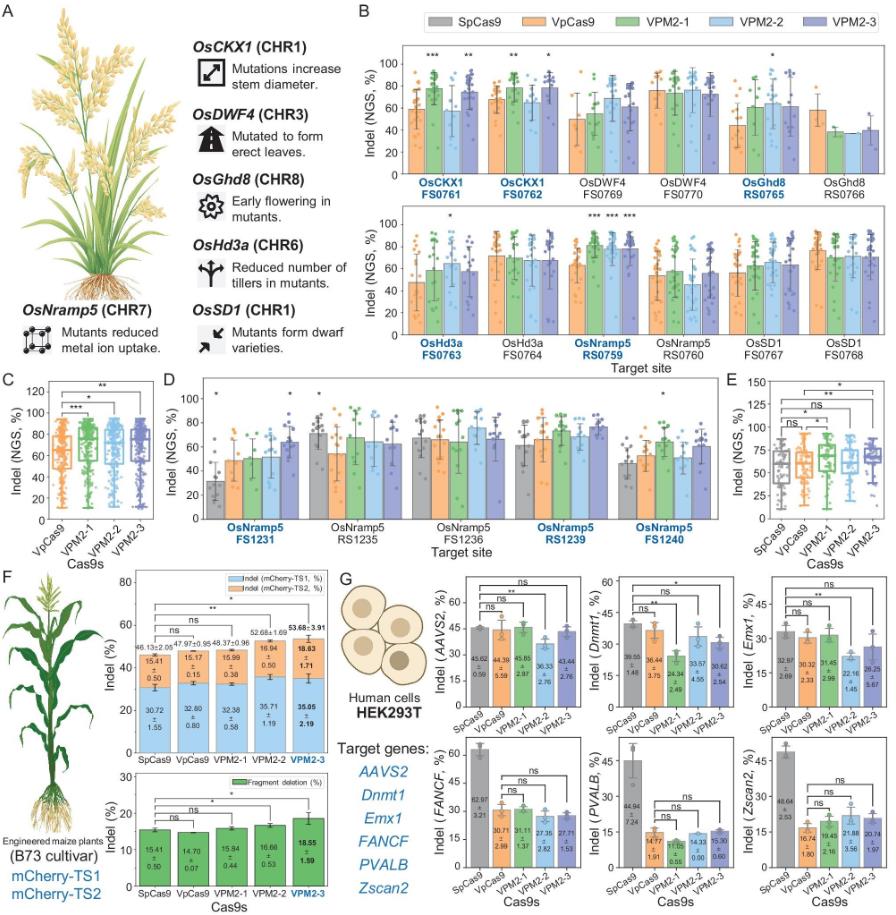
Figure 1.Application of VpCas9 and mutants for genome editing in rice callus, maize protoplasts and HEK293T cells. (Xu, et al. 2026))
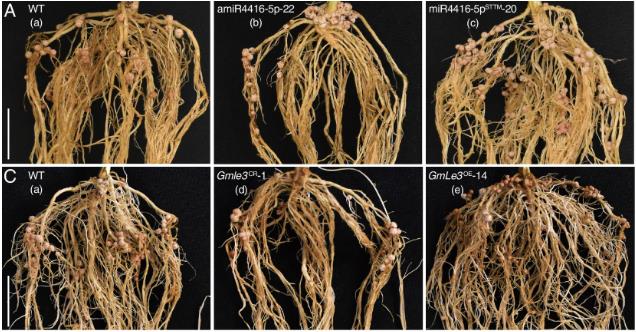
Twelve target sites across six genes (OsCKX1, OsDWF4, OsGhd8, OsHd3a, OsNramp5, OsSD1) were tested in rice callus:
Head-to-head comparison with SpCas9 at five additional OsNramp5 target sites showed that VPM2-1 (65.23%) and VPM2-3 (66.03%) significantly outperformed both VpCas9 (58.23%) and SpCas9 (56.06%). Off-target rates of all three VpCas9 mutants were comparable to SpCas9.
In stably transformed rice plants, overall editing efficiency was consistent with callus results, with VPM2-3 showing the highest average activity at 70.12%.
Two mCherry reporter target sites were tested:
Six genomic loci were tested in HEK293T cells. VpCas9 and its mutants edited all targets. At AAVS2, Dnmt1, and Emx1, efficiencies matched SpCas9, while at FANCF, PVALB, and Zscan2, they fell short. Importantly, off-target analysis revealed that SpCas9 produced substantial off-target editing at AAVS2_OTS_219 (51.31%) and FANCF_OTS_256 (23.57%), whereas VpCas9 and its mutants showed no detectable off-target events.
Systematic benchmarking against MP-TRANS, ESM2, random forest, and SVM classifiers confirmed CasMiner's superiority. The shuffling ratio proved to be a critical hyperparameter, with 80% emerging as optimal — neither too low (leaving residual features) nor too high (making classification trivially easy).
When compared with traditional homology tools:
The recommended practical workflow combines keyword and Pfam domain pre-filtering with CasMiner scoring, balancing computational cost and discovery power.
Despite these limitations, CasMiner establishes a proof of concept that deep learning can move beyond predicting the behavior of known CRISPR tools to discovering and engineering entirely new ones. For researchers working on plant genome editing, the demonstration that VPM2-3 outperforms SpCas9 in both rice and maize is particularly encouraging, suggesting that the Cas9 toolbox for crop improvement still has substantial room to expand.
Get Latest Lifeasible News and Updates Directly to Your Inbox