Crop improvement has always been, at its core, a story of DNA regulation. From ancient domestication to modern breeding, the traits that define higher yields, stress tolerance, and better nutritional profiles often trace back to changes in cis-regulatory DNA — the non-coding sequences that control when, where, and how much a gene is expressed. Yet despite their central role, the sequence-to-function relationships of these regulatory elements remain poorly understood. A landmark study published in Nature Biotechnology by Groover, Ding, Wang, Benegas, and colleagues now brings unprecedented clarity to this problem, using a high-throughput functional genomics platform in sorghum to systematically map how tens of thousands of cis-regulatory mutations influence the expression of three key photosynthesis genes.
CRISPR-based genome editing has opened the door to a strategy the authors call quantitative trait engineering (QTE) — the precise modification of cis-regulatory elements to dial gene expression up or down without introducing foreign DNA. This approach is particularly attractive for two reasons. First, it sidesteps the regulatory hurdles and public skepticism that accompany transgenic overexpression. Second, it offers a way to fine-tune endogenous gene expression rather than relying on exogenous promoters and enhancers, which can cause ectopic or excessive expression.
The challenge, however, is that hypermorphic mutations — those that increase gene expression — are rare. In a recent QTE screen targeting the rice PsbS gene, only 2 out of 120 mutated promoters yielded high-expression alleles. Without a systematic understanding of which non-coding positions matter and how they function, cis-regulatory editing remains largely a guessing game.
To address this knowledge gap, the researchers developed a massively parallel reporter assay (MPRA) in sorghum (Sorghum bicolor cv. RTx430) protoplasts. The workflow is elegant in its simplicity and power:
A critical design choice distinguishes this work from prior MPRA studies: rather than using minimal promoter fragments, the libraries retain the full native 2-kb 5' promoter and 5' UTR context, preserving complete gene structure. Each library contains approximately 10,000 mutations spanning the 2-kb promoter region and 5' UTR, and the mutations fall into three categories that directly mirror CRISPR editing outcomes:
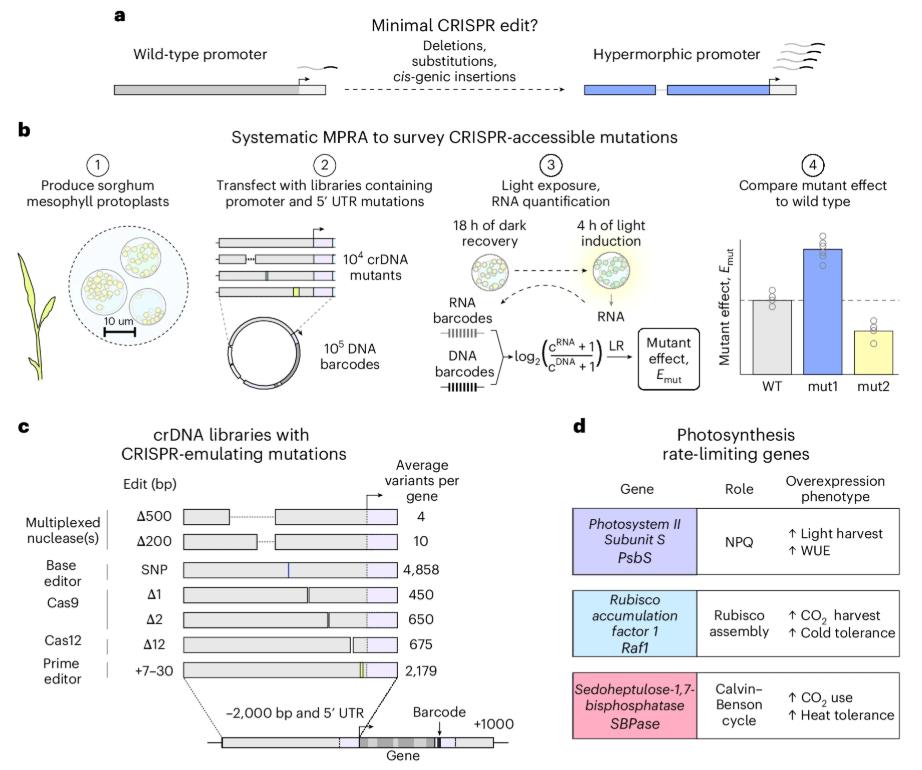
Figure 1. An MPRA for investigating CRISPR editing outcomes on sorghum cis-regulation. (Groover, et al. 2026)
The study focused on three photosynthesis genes with distinct biological roles and expression patterns:
This diversity of expression profiles makes the three genes an ideal test set for understanding whether cis-regulatory logic is universal or gene-specific.
Analysis of 10,096 PsbS mutations revealed a clear functional architecture. A compact core promoter region spanning approximately −400 bp to the translational start site (roughly 500 bp in total) harbors the vast majority of variants with large expression effects. In contrast, the distal promoter region (beyond -400 bp) shows much smaller mutational effect variance, consistent with chromatin accessibility dropping at approximately 400 bp upstream of the transcription start site.
Reproducibility mirrors this pattern: biological replicates correlate well within the core promoter (Pearson r = 0.68) but poorly in the distal region (r = 0.30). Within the core promoter, deletions are particularly reproducible (r = 0.75), while insertions are somewhat noisier (r = 0.62).
Validation with 12 promoter and 5' UTR variants confirmed that MPRA effect sizes correlate strongly with nanoluciferase protein output (r = 0.80). Importantly, when the same mutations were tested in a synthetic GFP construct, the correlation disappears (r = -0.26), demonstrating that the mutational effects depend on the native gene structure and cannot be captured by minimal promoter assays.
Within the core promoter, two classes of regulatory hotspots emerge:
Hypomorphic (expression-reducing) deletions cluster in a narrow region from -180 to -120 bp, indicating a core transcriptional function. Even a 12-bp deletion at position -168 reduces expression more than a 201-bp deletion at -200, underscoring the functional density of this region. These hypomorphic deletions overlap deeply conserved non-coding sequences found across related grass species, predating the C3-to-C4 evolutionary transition.
Hypermorphic (expression-increasing) deletions distribute across two zones:
Single-nucleotide variants and small deletions at positions -169 and -134 abolish PsbS expression entirely. These sites overlap conserved antisense-strand G-box and I-box motifs, which are known to be essential for light-mediated activation of photosynthetic genes in C4 plants. Intriguingly, while these motifs appear multiple times in the PsbS upstream region, mutations at other instances do not affect expression, highlighting the position-dependent nature of cis-regulatory function.
Not all motif changes are deleterious. A C-to-T conversion at position -133 modifies a native I-box-like sequence (GATAGGG) to a more canonical GATAAGG, producing an activating effect. This illustrates how subtle sequence changes can shift the affinity of transcription factor binding sites.
The -110 to -70 hypermorphic deletion zone overlaps C4-specific Myb/bZIP transcription factor binding sites (CAGTTG) and CAT box elements (GCCACT), suggesting that overexpression may arise from the removal of cell-type-specific repressive modules that evolved during C4 photosynthesis.
Validation in light-grown rice protoplasts confirmed the translational relevance of these findings: key PsbS mutations showed protein production levels correlated with sorghum MPRA values (r = 0.59) and with sorghum protein output (r = 0.89). Consistent with these MPRA results, a prior in planta promoter mutagenesis study in rice reported that G-box deletion reduced NPQ, while combined G-box and I-box deletion phenocopied a PsbS knockout, supporting that MPRA measurements can translate to whole-plant phenotypes.
Beyond deletions, the study tested 80 distinct cis-regulatory motifs (8–25 bp) inserted at 5-bp intervals across the -150 to +45 region, in both forward and reverse orientations. Eleven specific insertions produced significant overexpression after Bonferroni correction.
Key findings include:
Strikingly, compact deletions in the native promoter outperformed the expression boost provided by an industrial-standard transgenic enhancer at the tested position, further strengthening the case for cis-regulatory editing as a viable alternative to transgenic overexpression.
A major conclusion of the study is that cis-regulatory architecture is not one-size-fits-all. Analysis of 7,796 Raf1 mutations and 13,974 SBPase mutations revealed that while both genes also show elevated effect variance in a core promoter region starting around -400 bp, the specific regulatory structures differ substantially:
Motif insertion positional dependencies also vary:
The types of mutations that produce overexpression differ by gene:
For SBPase, 35 unique motifs across 74 insertion instances produced significant overexpression, with roughly two-thirds containing AGTCAA or GGATAA (I-box-like) sub-motifs. The top SBPase insertions (a 10-bp insertion at +30 and a 15-bp dual I-box insertion at -45) exceeded 16-fold overexpression, surpassing the 3×ENH enhancer at approximately 8-fold.
These results underscore a critical practical implication — that effective QTE requires gene-specific cis-regulatory maps. A mutation strategy that works for one gene may fail entirely for another.
The researchers fine-tuned a genomic pretrained network (GPN) — a genome-scale language model — to predict RNA-seq expression levels from sorghum leaf tissue promoter sequences (-256 to +256 bp). For PsbS, the model achieved moderate correlation between predicted and observed mutational effects in the core promoter (r = 0.51). Performance improved for medium-sized deletions of 10–15 bp (r = 0.76) but dropped for motif insertions (r = 0.39), and the model struggled to identify hypermorphic variants.
For Raf1 and SBPase, predictions were substantially weaker, likely because these genes are expressed at low levels in the tissue used for training, limiting the signal available to the model. In silico mutagenesis effects from the model matched measured single-nucleotide variant effects, but the model is best at predicting expression decreases rather than expression increases.
The practical consequence of this asymmetry is that computational models alone remain insufficient for discovering high-expression alleles. MPRA-based experimental screening remains essential for identifying the rare hypermorphic mutations that are most valuable for crop improvement.
This study provides the most comprehensive functional map of cis-regulatory variation in a crop species to date, offering several takeaways for researchers working in plant gene editing and functional genomics:
Several limitations deserve attention. MPRA results need validation in intact sorghum plants, and protoplast systems can only capture tissue-independent and condition-independent regulatory processes. Regulatory landscapes may differ in whole-plant contexts where cell-cell signaling and developmental programs shape gene expression. Additionally, the regulatory status of insertions versus deletions varies across countries, a practical consideration for translating QTE into commercial cultivars.
Looking ahead, expanding this approach to additional genes, tissue types, and stress conditions will build the comprehensive cis-regulatory atlases needed to make quantitative trait engineering a routine tool in crop improvement. The combination of high-throughput functional assays, precise CRISPR editing, and increasingly capable predictive models points toward a future where tuning gene expression is as deliberate and predictable as editing coding sequences — but without the regulatory burden of transgenes.
Get Latest Lifeasible News and Updates Directly to Your Inbox