CRISPR-Cas9 genome-wide knockout screening is a core tool in functional genomics research. By systematically perturbing genes and observing phenotypic changes, researchers can reveal gene function on a large scale. However, this technology has always faced two mutually constraining challenges: guide RNAs may not be able to effectively target the target gene (insufficient on-target efficacy), and may also produce off-target effects at non-target sites (off-target activity).
These two problems have spurred numerous prediction algorithms and design strategies, but for a long time, a unified framework that can simultaneously balance both has been lacking. Meanwhile, with the widespread adoption of high-dimensional readout technologies (such as single-cell RNA sequencing and high-content imaging), researchers' demand for more compact and efficient screening libraries is increasing, while the continuous updating of genome annotation is gradually rendering existing libraries obsolete.
On March 25, 2026, John G. Doench's team from the Broad Institute of MIT and Harvard proposed a systematic solution in a paper—they designed and validated novel whole-genome CRISPR-Cas9 knockout libraries Jacquere (human) and Julianna (mouse), and developed a new strategy, CRISPick aggregate CFD, to balance on-target efficacy and off-target avoidance.
To construct an excellent library, a precise understanding of the patterns of off-target activity is essential. Using GUIDE-seq data from 114 unique guides, the research team systematically quantified the activity rate of off-target sites in CD4+/CD8+ T cells, U2OS, and HEK293 cell lines:
This result clearly shows that off-target activity decreases sharply with increasing mismatch numbers. Notably, alignment shifts in RNA/DNA bulges do not increase activity, a finding that simplifies the dimensions considered in off-target assessment.
Further analysis revealed a high correlation between the CFD (cutting frequency determination) score and the activity rate measured by GUIDE-seq (Pearson R = 0.93), providing a reliable basis for assessing off-target risk based on computational prediction.
Based on the above findings, the research team proposed the CRISPick aggregate CFD classifier. The core idea is to represent each guide as the sum of the CFD scores of all its off-target sites, and to distinguish between guides with high and low specificity by setting a threshold.
A key question is: how many mismatched off-target sites should be included? The team evaluated the classifier's F1 score at several mismatch thresholds:
Including too many mismatched off-target sites introduces a large amount of noise from inactive sites, reducing classifier performance. The optimal approach is to consider only off-target sites with at most 1 mismatch in the SDRs, with a threshold set at 4.8.
Validation on the Avana library dataset shows that the classifier's performance is unaffected by TP53 status—the F1 score is 0.73 for wild-type TP53 and 0.72 for mutant TP53.
The study also compared the widely used GuideScan specificity score. Because GuideScan included too many inactive off-target sites (87.9% of the included sites were inactive, compared to only 56.3% in CRISPick aggregate CFD), it performed significantly worse. This further confirms an important principle: not all alignable off-target sites are worth paying attention to; over-inclusion only dilutes the signal.
Guided by the CRISPick aggregate CFD classifier, the research team designed a novel human whole-genome CRISPR-Cas9 knockout library, Jacquere. Its design process reflects multiple considerations:
The entire library contains 60,550 unique guides, of which 95.0% were selected in the first round of selection, indicating that most genes had highly active and specific guides available.
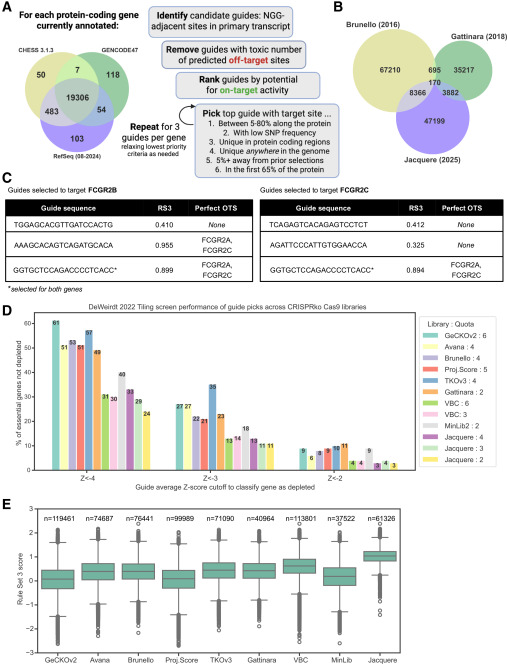
Figure 1. Composition of Jacquere and comparison across CRISPR-Cas9ko genome-wide libraries. (Drepanos, et al. 2026)
The Jacquere library outperforms existing mainstream libraries in several dimensions:
The research team performed depletion screen validation in A549 (lung cancer) and A375 (melanoma) cell lines. Good consistency was observed between biological replicates (Pearson r = 0.93 and 0.95, respectively).
Compared to the widely used Brunello library, Jacquere showed significant advantages:
More importantly, the false negative rate was significantly reduced. Of the 131 essential genes that Brunello failed to detect, Jacquere recovered 97, with a similar false positive rate.
The study also compared the performance of single-guide and dual-guide vectors. Dual-guide vectors introduced more false positives—the depletion rate of non-essential genes was as high as 31.1% in Vienna-dual and 18.0% in Vienna-single, while Jacquere only reached 2.7%. This result suggests that while pursuing higher screening efficiency, the dual-guide strategy may introduce additional noise, requiring careful trade-offs.
A well-designed library paired with an inappropriate analysis method can still produce misleading results. The research team compared three commonly used hit identification methods: Z-score, MAGeCK RRA, and MAGeCK MLE.
All three methods effectively distinguished between essential and non-essential genes, and performed better on Jacquere data. However, MAGeCK RRA has significant limitations: it relies solely on rank information while ignoring effect size, potentially leading to false positives (an anomaly in a single guide can cause a gene to be reported as a hit) and false negatives (when the depletion levels of different guides are inconsistent).
The research team provided a specific cautionary example: in an NK cell study, MAGeCK RRA reported Calhm2 as a positive selection hit, but none of its targeting guides were even among the top 25% of positively selected guides in the library. In such cases, results from a single method are unreliable.
Therefore, it is recommended to use multiple methods to cross-validate hits to avoid false findings due to systematic biases from a single method.
While the Jacquere library performed exceptionally well in design and validation, the research team candidly pointed out several limitations:
This work provides the CRISPR screening field with a rigorously validated, transparently designed whole-genome library. More importantly, it establishes a generalizable framework—finding a balance between on-target efficacy and off-target avoidance, making screening results more reliable.
Get Latest Lifeasible News and Updates Directly to Your Inbox